Ce este sindromul Ehlers-Danlos (EDS)
Sindromul Ehlers-Danlos (EDS) reprezintă un grup de 13 afecțiuni genetice rare ale țesutului conjunctiv, asociate cu hipermobilitate articulară, hiperextensibilitate a pielii și fragilitate tisulară.
Sindromul Ehlers-Danlos este cauzat de mutații ale genelor implicate în formarea colagenului și integritatea țesutului conjunctiv, conducând la apariția unui spectru larg de manifestări clinice de severitate variabilă precum:
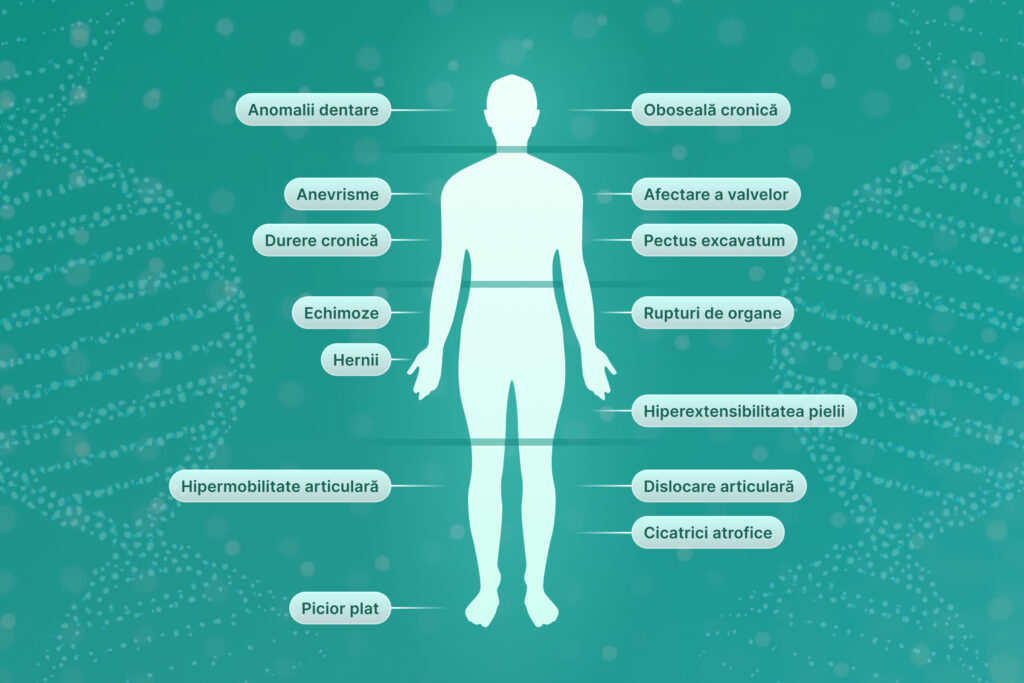
- cele articulare: hipermobilitate articulară, dislocații articulare repetate;
- cele de la nivelul pielii și țesuturilor conjunctive moi: hiperextensibilitate a pielii, formare a cicatricilor atrofice, hernii, rupturi de organe;
- cele vasculare: apariția hematoamelor la traumatisme minime, anevrismelor, disecțiilor;
- simptome generale: oboseală, dureri cronice, probleme dentare, probleme de vedere.
Cele 13 tipuri de EDS prezintă semne și simptome comune și particulare, ajutând la încadrarea clinică în tipul specific și orientarea către testarea genetică potrivită pentru diagnosticul de certitudine [1].
Importanța conștientizării și înțelegerii EDS
Impactul sindromului Ehlers-Danlos asupra pacienților poate fi profund, afectând calitatea vieții și exercitând presiuni emoționale și sociale considerabile. Persoanele cu EDS se confruntă adesea cu dureri cronice, dislocări frecvente ale articulațiilor și probleme dermatologice, ceea ce le limitează capacitatea de a desfășura activități zilnice obișnuite. În unele cazuri, cum ar fi în forma vasculară a EDS, există un risc crescut de rupturi ale vaselor de sânge și organelor interne, care pot pune viața în pericol.
Conștientizarea și înțelegerea acestei afecțiuni sunt esențiale pentru a facilita diagnosticul precoce și pentru a îmbunătăți gestionarea sa. Un diagnostic corect și precoce poate ajuta la implementarea unor măsuri de prevenție și monitorizare adecvate, cum ar fi monitorizarea cardiovasculară regulată și evitarea activităților care ar putea agrava simptomele. De asemenea, o mai bună înțelegere a EDS în rândul cadrelor medicale și al publicului larg poate contribui la reducerea întârzierilor în diagnostic și la îmbunătățirea calității vieții pacienților prin planuri de tratament adaptate nevoilor lor specifice. Mai mult, prin depistarea preconcepțională a acestui sindrom, persoana afectată poate opta pentru testarea genetică prenatală, cu scopul de a împiedica transmiterea afecțiunii copiilor săi, sau neonatală, cu scopul de a gestiona manifestările fizice timpuriu, din copilărie.
Înțelegerea sindromului Ehlers-Danlos
Context istoric și descoperire
Primele descrieri ale hipermobilității articulare datează din anul 400 î.Hr., când Hipocrate a menționat astfel de cazuri în scrierile sale. Însă, sindromul Ehlers-Danlos a fost recunoscut pentru prima dată ca o entitate distinctă de către dermatologul danez Edvard Ehlers în 1901, iar mai târziu, în 1908, Henri-Alexandre Danlos a adăugat detalii esențiale despre caracteristicile cutanate ale afecțiunii. Denumirea de “Sindrom Ehlers-Danlos” a fost oficializată în 1936, când s-a stabilit că această afecțiune cuprinde mai multe subtipuri cu manifestări comune [2].
Baza genetică a EDS
EDS este rezultatul mutațiilor care afectează genele responsabile pentru producerea și utilizarea colagenului.
Mutațiile pot fi transmise de la părinți purtători sănătoși în cazul tipurilor autozomal recesive de EDS precum tipul cifoscoliotic, pot fi transmise de la un părinte afectat sau chiar pot apărea de novo (primul caz din familie) în cazul tipurilor autozomal dominante precum cel vascular sau cel clasic.
Astfel, cunoscând mutația și gena implicate se poate estima riscul de a avea un copil afectat și opțiunile reproductive potrivite pentru stoparea transmiterii afecțiunii.
Diferențele dintre diferitele tipuri
Fiecare tip al sindromului Ehlers-Danlos prezintă simptome și riscuri distincte. De exemplu, EDS hipermobil (hEDS) afectează în principal articulațiile și pielea, ducând la dureri cronice și instabilitate articulară. În schimb, EDS vascular (vEDS) este dominat de riscuri vasculare, cu rupturi spontane ale arterelor și organelor interne, o consecință a mutațiilor care afectează colagenul de tip III [1][3]. Cunoașterea manifestărilor și riscurilor asociate fiecărui tip de sindrom Ehlers-Danlos poate ajuta la gestionarea eficientă a afecțiunii.
Simptomele și caracteristicile clinice ale sindromului Ehlers-Danlos
Simptome comune pentru toate tipurile EDS
Sindromul Ehlers-Danlos (EDS) cuprinde o serie de simptome comune, prezente la majoritatea subtipurilor:
- Hipermobilitate articulară: Persoanele cu EDS prezintă articulații extrem de flexibile, care permit o gamă mai largă de mișcări decât în mod obișnuit. Acest lucru poate conduce la dislocații frecvente și dureri articulare cronice. Hipermobilitatea poate afecta articulațiile mari (umerii, genunchii) și mici (degetele), compromițând stabilitatea acestora și provocând simptomatologie pe termen lung.
- Manifestări cutanate: Pielea persoanelor cu EDS este elastică și poate fi întinsă mai mult decât este normal. În unele tipuri de EDS precum cel clasic-like, pielea revine la forma inițială atunci când este eliberată, dar în alte tipuri, aceasta poate fi fragilă și predispusă la vânătăi și cicatrizare anormală precum în tipul dermatosparaxis.
- Fragilitatea țesuturilor: Țesuturile conjunctive fragile, cauzate de defectele de colagen, predispun pacienții cu EDS la vânătăi spontane, vindecare lentă și formarea de cicatrici subțiri și atrofice. De asemenea, pacienții pot suferi complicații severe precum rupturi de organe.
- Manifestări vasculare: Afectarea componentei conjunctive a peretelui vascular poate predispune persoanele cu EDS la manifestări vasculare severe precum disecții sau rupturi vasculare.
Simptome specifice tipului și manifestări
Fiecare tip de EDS prezintă simptome specifice, pe lângă manifestările comune majorității tipurilor. Astfel, EDS vascular (vEDS) este caracterizat prin riscuri majore de rupturi arteriale și a organelor interne. Pacienții cu vEDS au o piele foarte subțire, translucidă, prin care sunt vizibile vasele de sânge. De asemenea, aceștia pot avea trăsături faciale distinctive, cum ar fi un nas subțire, buze subțiri și loburi mici ale urechilor.
Iar tipul clasic de EDS (cEDS) este dominat de manifestări cutanate, precum pielea care se rupe cu ușurință la traumatisme minime și cicatrici subțiri, asemănătoare hârtiei de țesut. În plus, persoanele afectate pot prezenta pseudotumori molluscoide, mici noduli ce se formează pe cicatrici, și papule piezogenice, proeminențe adipoase care apar pe călcâie.
Complicații și manifestări asociate
Persoanele cu EDS pot experimenta o serie de complicații precum dislocațiile frecvente ale articulațiilor, prolapsul valvei mitrale și dilatarea rădăcinii aortice.
Aceste complicații necesită intervenții rapide sau monitorizări constante precum cea cardiologică, pentru gestionarea imediată a complicației sau prevenirea altor evenimente.
Complicațiile asociate EDS, sunt frecvent asociate cu sindromul tahicardiei ortostatice posturale (POTS) și de sindromul de activare mastocitară (MCAS).
POTS se caracterizează printr-o creștere anormală a ritmului cardiac la ridicarea în picioare, în timp ce MCAS presupune episoade de reacții alergice severe și neobișnuite. Conexiunea dintre EDS, POTS și MCAS este din ce în ce mai recunoscută, prevalența acestor afecțiuni fiind mai mare la pacienții cu EDS comparativ cu populația generală.
Uneori, persoanele cu EDS pot acuza ca principale supărări, simptome nespecifice precum oboseala cronică, amețeala, dureri de cap, tulburări gastrointestinale.
Înțelegerea acestor simptome și caracteristici clinice este esențială pentru a facilita diagnosticul și gestionarea sindromului Ehlers-Danlos, deoarece manifestările pot varia considerabil între tipuri și necesită abordări terapeutice personalizate.
Diagnosticul sindromului Ehlers-Danlos
Evaluarea clinică și criteriile de diagnostic
Diagnosticarea sindromului Ehlers-Danlos (EDS) începe cu o evaluare medicală detaliată, care implică analiza istoricului medical al pacientului, istoricul familial și un examen fizic amănunțit. Caracteristici esențiale precum hipermobilitatea articulară, hiperextensibilitatea cutanată și fragilitatea țesuturilor sunt examinate în detaliu. Consemnarea acestor semne și simptome clinice este utilizată pentru a ghida diagnosticul, oferind astfel o bază solidă pentru identificarea corectă a afecțiunii.
Criterii pentru diagnosticarea diferitelor tipuri de EDS
Fiecare tip de EDS are criterii specifice de diagnostic, care includ caracteristici clinice majore și minore. Aceste criterii ajută la diferențierea celor 13 tipuri recunoscute. De exemplu, diagnosticul EDS clasic (cEDS) se bazează pe prezența hiperextensibilității cutanate și a cicatricilor atrofice, în timp ce EDS vascular (vEDS) se distinge prin riscul de ruptură arterială și prezența pielii subțiri. În cazul EDS hipermobil (hEDS), diagnosticul este clinic, deoarece în prezent nu există un test genetic specific pentru acest tip.
Provocări în diagnosticarea EDS
Variabilitatea clinică largă a simptomelor
Diagnosticarea EDS poate fi dificilă din cauza variabilității largi a simptomelor și a suprapunerilor acestora cu alte tulburări ale țesutului conjunctiv. Natura subiectivă a unor caracteristici clinice, precum hipermobilitatea articulară și textura pielii, poate crea confuzii și poate duce la întârzierea diagnosticării corecte.
Necesitatea unei abordări multidisciplinare
Datorită complexității manifestărilor sindromului Ehlers-Danlos, diagnosticarea și gestionarea corectă necesită o abordare multidisciplinară. Acest lucru presupune colaborarea între geneticieni, ortopezi, dermatologi, cardiologi și alți specialiști care pot evalua și gestiona diversele complicații asociate cu EDS. O abordare multidisciplinară asigură o îngrijire holistică și optimizează rezultatele pentru pacienți, tratând atât simptomele imediate, cât și prevenind complicațiile viitoare.
Testarea genetică și diagnosticul molecular
Rolul testării genetice în confirmarea EDS
Testarea genetică are un rol esențial în confirmarea diagnosticului pentru majoritatea tipurilor de EDS prin identificarea mutațiilor în genele specifice care sunt implicate în sinteza și utilizarea colagenului și integritatea țesutului conjunctiv. Acest aspect este deosebit de important pentru tipuri precum vEDS, unde mutațiile în gena COL3A1 pot confirma diagnosticul și pot ghida tratamentul și monitorizarea specifice. Testarea genetică este esențială pentru a stabili diagnosticul de certitudine de EDS și pentru a exclude alte afecțiuni cu manifestări similare.
Progresele în diagnosticul molecular și ce înseamnă acestea pentru pacienții cu EDS
Tehnicile moderne de diagnostic molecular, cum ar fi secvențierea de nouă generație (NGS), au revoluționat capacitatea de a detecta mutațiile genice care confirmă diagnosticul de EDS. Aceste tehnologii permit secvențierea simultană a mai multor gene, oferind un diagnostic mai rapid și mai precis. Aceste progrese sunt extrem de benefice pentru pacienți, deoarece scurtează timpul până la diagnostic și permit stabilirea unor planuri de gestionare adaptate tipului specific de EDS și monitorizarea mai eficientă pentru prevenirea complicațiilor.
Clasificarea clinică a EDS:
În 2017, s-a stabilit o clasificare oficială a sindromului Ehlers-Danlos, recunoscând 13 subtipuri distincte, fiecare cu un set de criterii clinice și mutații genetice specifice [1].
EDS clasic (cEDS)
Mod de transmitere: autozomal dominant
Genele implicate: COL1A1, COL5A1, COL5A2
Pentru diagnosticul clinic de cEDS sunt necesare un criteriu major și minim 3 criterii minore sau două criterii majore.
Criterii majore:
- Hiperextensibilitatea pielii și cicatrizarea atrofică
- Hipermobilitatea generalizată a articulațiilor
Criterii minore:
- Piele ușor susceptibilă la echimoze
- Piele moale, de consistența aluatului
- Fragilitate cutanată (sau fisurare traumatică)
- Pseudotumori molluscoide
- Sferoizi subcutanați
- Hernie în antecedente
- Epicantus
- Complicații ale hipermobilității articulare (de ex., entorse, luxații/subluxații, durere, picior plat flexibil)
- Istoric familial cu rude de gradul I care îndeplinesc criteriile clinice
EDS clasic-like (clEDS)
Mod de transmitere: autozomal recesiv
Gena implicată: TNXB
Pentru diagnosticul clinic de clEDS sunt necesare 3 criterii majore și un istoric familial sugestiv pentru modul de transmitere autozomal recesiv
Criterii majore:
- Hiperextensibilitatea pielii, cu o textură catifelată a acesteia și absența cicatricelor atrofice
- Hipermobilitate articulară generalizată, cu sau fără dislocări recurente (cel mai frecvent ale umărului și gleznei)
- Piele ușor susceptibilă la echimoze
Criterii minore:
- Deformități ale piciorului: antepicior lat și plin, brahidactilie cu exces de piele; picior plat; hallux valgus; papule piezogene
- Edem la nivelul membrelor inferioare în absența insuficienței cardiace
- Slăbiciune musculară ușoară proximală și distală
- Polineuropatie axonală
- Atrofie musculară la mâini și picioare
- Mâini acrogerice, halux ciocan, clinodactilie, brahidactilie
- Prolaps vaginal/uterin/rectal
EDS cardiac-valvular (cvEDS)
Mod de transmitere: autozomal recesiv
Gena implicată: COL1A2
Pentru diagnosticul clinic de cvEDS sunt necesare criteriul major: probleme cardiace-valvulare severe progresive + un alt criteriu major sau cel puțin două criterii minore și un istoric familial sugestiv pentru transmitere autozomal recesivă
Criterii majore:
- Probleme cardiace-valvulare severe progresive (valva aortică, valva mitrală)
- Implicarea pielii: hiperextensibilitatea pielii, cicatrici atrofice, piele subțire, echimoze la traumatisme minime
- Hiperlaxitate articulară (generalizată sau localizată la articulații mici)
Criterii minore:
- Hernie inghinală
- Deformare a toracelui (în special pectus excavatum)
- Luxații articulare
- Deformări ale piciorului: picior plat (pes planus), picior planovalg (pes planovalgus), hallux valgus
EDS vascular (vEDS)
Mod de transmitere: autozomal dominant
Genele implicate: COL1A1, COL3A1
Testarea genetică este recomandată în cazul unui istoric familial pozitiv pentru această afecțiune sau manifestări clinice sugestive pentru această afecțiune, o ruptură sau disecție arterială la persoane cu vârsta sub 40 de ani, perforația inexplicabilă a colonului sigmoid sau pneumotorax spontan în prezența altor caracteristici compatibile cu vEDS ar trebui să conducă la investigații de diagnostic. Testarea pentru vEDS ar trebui luată în considerare în prezența unei combinații de alte caracteristici clinice minore enumerate mai jos.
Criterii majore:
- Istoric familial de vEDS cu variantă cauzatoare documentată în COL3A1
- Ruptură arterială la o vârstă tânără
- Perforație spontană a colonului sigmoid în absența unei boli diverticulare cunoscute sau a altor patologii intestinale
- Ruptură uterină în timpul celui de-al treilea trimestru, în absența unei cezariene anterioare și/sau a unor rupturi perineale severe peripartum
- Formarea fistulei carotido-cavernoase (CCSF) în absența unui traumatism
Criterii minore:
- Echimoze neasociate cu un traumatism identificat și/sau în zone neobișnuite precum obrajii și spatele
- Piele subțire, translucidă, cu vizibilitate crescută a venelor
- Aspect facial caracteristic: hipertelorism (ochi îndepărtați), ochi proeminenți, buze subțiri, micrognație (bărbie mică, situată înapoi), nas îngust, obraji înfundați
- Pneumotorax spontan
- Acrogerie (îmbătrânire prematură a pielii)
- Talipes equinovarus
- Luxație congenitală a șoldului
- Hipermobilitate a articulațiilor mici
- Ruptură de tendoane și mușchi
- Keratoconus
- Recesie gingivală și fragilitate gingivală
- Varice cu debut precoce (sub 30 de ani și la femei nulipare)
EDS hipermobil (hEDS)
Mod de transmitere: autozomal dominant
Genele implicate: necunoscute până în prezent
Pentru diagnosticul clinic este necesară îndeplinirea a celor 3 criterii:
Criteriul 1: Hipermobilitate articulară generalizată
Criteriul 2: Două sau mai multe dintre următoarele caracteristici (A–C) TREBUIE să fie prezente:
Caracteristica A: manifestări sistemice ale unei afecțiuni generalizate a țesutului conjunctiv (trebuie să fie prezente un total de cinci)
- Piele neobișnuit de moale sau catifelată
- Hiperextensibilitate cutanată ușoară
- Striae neexplicate, cum ar fi striae distensae sau rubrae pe spate, inghinal, coapse, sâni și/sau abdomen la adolescenți, bărbați sau femei prepubertare fără un istoric de creștere sau pierdere semnificativă de grăsime corporală
- Papule piezogene bilaterale ale călcâiului
- Hernii abdominale recurente sau multiple (de exemplu, ombilicale, inghinale, crurale)
- Cicatrici atrofice care implică cel puțin două locuri și fără formarea cicatricilor cu aspect de hârtie și/sau largi, așa cum se observă în EDS clasic
- Prolaps pelvian, rectal și/sau uterin la copii, bărbați sau femei nulipare fără un istoric de obezitate morbidă sau altă afecțiune medicală predispozantă cunoscută
- Înghesuire dentară și palat înalt sau îngust
- Arahnodactilie, definită prin una sau mai multe dintre următoarele: (i) semnul pozitiv al încheieturii (semnul Steinberg) bilateral; (ii) semnul pozitiv al degetului mare (semnul Walker) bilateral
- Anvergura brațelor raportat la înălțime ≥1.05
- Prolaps de valvă mitrală (MVP) ușor sau mai sever, conform criteriilor ecocardiografice stricte
- Dilatarea rădăcinii aortei cu un scor Z > +2
Caracteristica B: istoric familial pozitiv, cu unul sau mai multe rude de gradul întâi care îndeplinesc independent criteriile de diagnostic curente pentru hEDS.
Caracteristica C: complicații musculo-scheletice (trebuie să fie prezentă cel puțin una)
- Durere musculo-scheletică în două sau mai multe membre, recurentă zilnic timp de cel puțin 3 luni
- Durere cronică, răspândită, timp de ≥3 luni
- Luxații articulare recurente sau instabilitate articulară evidentă, în absența unui traumatism (a sau b)
- Trei sau mai multe luxații atraumatice în aceeași articulație sau două sau mai multe luxații atraumatice în două articulații diferite care apar la momente diferite
- Confirmarea medicală a instabilității articulare în două sau mai multe locuri, fără legătură cu traumatisme.
Criteriul 3: toate următoarele trebuiesc îndeplinite:
- Absența fragilității neobișnuite a pielii, care ar trebui să determine luarea în considerare a altor tipuri de EDS.
- Excluderea altor tulburări ereditare și dobândite ale țesutului conjunctiv, inclusiv afecțiuni reumatologice autoimune. La pacienții cu o tulburare dobândită a țesutului conjunctiv (de exemplu, lupus, artrită reumatoidă etc.), un diagnostic suplimentar de hEDS necesită îndeplinirea ambelor Caracteristici A și B ale Criteriului 2. Caracteristica C a Criteriului 2 (durere cronică și/sau instabilitate) nu poate fi luată în considerare pentru diagnosticarea hEDS în această situație.
- Excluderea diagnosticelor alternative care pot include și hipermobilitatea articulară prin hipotonie și/sau laxitate a țesutului conjunctiv. Diagnosticele și categoriile diagnostice alternative includ, dar nu se limitează la, tulburări neuromusculare (de exemplu, EDS miopatic, miopatie Bethlem), alte HCTD (de exemplu, alte tipuri de EDS, sindromul Loeys-Dietz, sindromul Marfan) și displazii scheletale (de exemplu, osteogenesis imperfecta). Excluderea acestor considerații poate fi bazată pe istoric, examen fizic și/sau testare genetică moleculară, după caz.
EDS artrochalazic (aEDS)
Mod de transmitere: autozomal dominant
Genele implicate: COL1A1, COL1A2
Pentru diagnosticul clinic al aEDS sunt necesare:
- 2 criterii majore: luxație congenitală de șold bilaterală și hiperextensibilitatea pielii sau
- 2 criterii majore luxație congenitală de șold bilaterală și hipermobilitate generalizată articulară severă cu multiple dislocări/subluxații și cel puțin 2 criterii minore
Criterii majore:
- Luxație congenitală bilaterală a șoldului
- Hipermobilitate articulară generalizată severă, cu multiple dislocări/subluxații
- Hiperextensibilitate cutanată
Criterii minore:
- Hipotonie musculară
- Cifoscolioză
- Osteopenie ușoară vizibilă radiologic
- Fragilitate tisulară, inclusiv cicatrici atrofice
- Piele ușor vulnerabilă la echimoze
EDS dermatospraxis (dEDS)
Mod de transmitere: autozomal recesiv
Genele implicate: ADAMTS2
Pentru diagnosticul clinic al dEDS sunt necesare 2 criterii majore: fragilitate extremă a pielii și trăsături craniofaciale caracteristice și un alt criteriu major și/sau trei criterii minore.
Criterii majore:
- Fragilitate extremă a pielii, cu rupturi cutanate congenitale sau postnatale
- Trăsături craniofaciale caracteristice, care sunt evidente la naștere sau în copilăria timpurie, sau evoluează mai târziu în copilărie
- Piele redundantă, aproape laxă, cu pliuri excesive la încheieturi și glezne
- Încrețire palmară accentuată
- Tendință severă de a dezvolta echimoze, cu risc de hematoame subcutanate și hemoragii
- Hernie ombilicală
- Întârziere a creșterii postnatale
- Membre, mâini și picioare scurte
- Complicații perinatale din cauza fragilității țesutului conjunctiv
Criterii minore:
- Textură moale a pielii, de aluat
- Hiperextensibilitatea pielii
- Cicatrici atrofice
- Hiperlaxitate articulară generalizată (GJH)
- Complicații ale fragilității viscerale (de exemplu, ruperea vezicii urinare, ruperea diafragmei, prolaps rectal)
- Întârziere în dezvoltarea motorie
- Osteopenie
- Hirsutism
- Anomalii dentare
- Erori de refracție (miopie, astigmatism)
- Strabism
EDS cifoscoliotic (kEDS)
Mod de transmitere: autozomal recesiv
Genele implicate: PLOD1, FKBP14
Pentru diagnosticul clinic al kEDS sunt necesare 2 criterii majore: hipotonie musculară congenitală și cifoscolioză congenitală sau cu debut devreme în copilărie și încă un criteriu major și/sau trei criterii minore.
Criterii majore:
- Hipotonie musculară congenitală
- Cifoscolioză congenitală sau cu debut timpuriu (progresivă sau neprogresivă)
- Hiperlaxitate articulară generalizată cu dislocări/subluxații (în special la umeri, șolduri și genunchi)
Criterii minore:
- Hiperextensibilitate cutanată
- Piele predispusă la echimoze
- Ruptură/anevrism al unei artere de mărime medie
- Osteopenie/osteoporoză
- Sclere albastre
- Hernie (ombilicală sau inghinală)
- Deformitate toracică (pectus excavatum)
- Habitus marfanoid
- Talipes equinovarus
- Erori de refracție (miopie, hipermetropie)
Criterii minore specifice genei PLOD1:
- Fragilitate a pielii (echimoze frecvente la traumatisme minime, piele friabilă, vindecare lentă a rănilor), cicatrici atrofice extinse
- Fragilitate/ruptură sclerală și oculară
- Microcornee
- Dismorfism cranio-facial
Criterii minore specifice genei FKBP14:
- Deficit auditiv congenital (neurosenzorial, conductiv sau mixt)
- Hiperkeratoză foliculară
- Atrofie musculară
- Diverticule vezicale
Sindromul corneei fragile (BCS)
Mod de transmitere: autozomal recesiv
Genele implicate: ZNF469, PRDM5
Pentru diagnosticul clinic al BCS sunt necesare un criteriu major: cornee subțire, cu sau fără ruptură și cel puțin un alt criteriu major și/sau trei criterii minore.
Criterii majore:
- Cornee subțire, cu sau fără ruptură (grosimea centrală a corneei adesea <400 µm)
- Debut timpuriu de keratoconus progresiv
- Debut timpuriu de keratoglobus progresiv
- Sclere albastre
Criterii minore:
- Enucleere sau cicatrici corneene ca rezultat al unei rupturi anterioare
- Pierderea progresivă a grosimii stromale corneene, în special în zona centrală a corneei
- Miopie mare, cu lungime axială normală sau moderat crescută
- Dezlipire de retină
- Surditate, adesea cu componente conductive și neurosenzoriale mixte, progresivă, frecvențele înalte fiind adesea mai grav afectate (audiogramă tonală pură „în pantă”)
- Membrane timpanice hipercompliabile
- Displazie de dezvoltare a șoldului
- Hipotonie în copilărie, de obicei ușoară, dacă este prezentă
- Scolioză
- Arahnodactilie
- Hipermobilitate a articulațiilor distale
- Platfus, hallux valgus
- Contracturi ușoare ale degetelor (în special al 5-lea)
- Piele moale, catifelată, translucidă
EDS spondilodisplastic (spEDS)
Mod de transmitere: autozomal recesiv
Genele implicate: B4GALT7, B3GALT6, SLC39A13
Pentru diagnosticul clinic al spEDS sunt necesare 2 criterii majore: statura mică și hipotonia musculară, plus anomalii radiografice și cel puțin alte 3 criterii minore generale sau specifice genei implicate.
Criterii majore:
- Statură mică (progresivă în copilărie)
- Hipotonie musculară (variind de la severă congenitală până la ușoară, cu debut tardiv)
- Îndoirea membrelor
Criterii minore:
- Hiperextensibilitatea pielii, piele moale, păstoasă, subțire și translucidă
- Platfus (pes planus)
- Dezvoltare motorie întârziată
- Osteopenie
- Dezvoltare cognitivă întârziată
Criterii minore specifice genei B4GALT7:
- Sinostoză radioulnară
- Contracturi bilaterale ale cotului sau mișcări limitate ale cotului
- Hiperlaxitate articulară generalizată
- Pliuri palmare transversale unice
- Trăsături craniofaciale caracteristice
- Constatări radiografice caracteristice
- Hipermetropie severă
- Cornee înnorată
Criterii minore specifice genei B3GALT6:
- Cifoscolioză (congenitală sau cu debut precoce, progresivă)
- Hipermobilitate articulară, generalizată sau restricționată la articulațiile distale, cu dislocări articulare
- Contracturi articulare (congenitale sau progresive) (în special la mâini)
- Degete particulare (subțiri, alungite, arahnodactilie, spatulate, cu falange distale late)
- Talipes equinovarus
- Trăsături craniofaciale caracteristice
- Decolorarea dinților, dinți displazici
- Constatări radiografice caracteristice
- Osteoporoză cu fracturi spontane multiple
- Anevrism de aortă ascendentă
- Hipoplazia plămânilor, boală pulmonară restrictivă
Criterii minore specifice genei SLC39A13:
- Ochi proeminenți cu scleră albăstruie
- Mâini cu palme fin ridate
- Atrofie a mușchilor thenari și degete alungite
- Hipermobilitatea articulațiilor distale
- Constatări radiologice caracteristice
EDS musculocontractural (mcEDS)
Mod de transmitere: autozomal recesiv
Genele implicate: CHST14, DSE
Pentru diagnosticul clinic al mcEDS sunt necesare:
- la naștere și în copilărie – 2 criterii majore: contracturi multiple congenitale și caracteristici cranio-faciale specifice
- în adolescență și la maturitate – 2 criterii majore: contracturi multiple congenitale și caracteristici cutanate specifice
Criterii majore:
- Contracturi multiple congenitale, caracteristic contracturi de adducție-flexie și/sau talipes equinovarus (picior strâmb)
- Caracteristici cranio-faciale specifice, evidente la naștere sau în primele luni de viață
- Caracteristici cutanate specifice, inclusiv hiperextensibilitate a pielii, tendință crescută la echimoze, fragilitate a pielii cu cicatrici atrofice, riduri palmare pronunțate
Criterii minore:
- Dislocații recurente/cronice
- Deformări ale toracelui/sternului
- Deformări ale coloanei vertebrale (scolioză, cifoscolioză)
- Degete neobișnuite (subțiri, cilindrice)
- Deformări progresive ale piciorului (valgus, planus, cavum)
- Hematoame subcutanate mari
- Constipație cronică
- Diverticuli colonici
- Pneumotorax/pneumohemotorax
- Nefrolitiază/cistolitiază
- Hidronefroză
- Criptorhidism la bărbați
- Strabism
- Erori de refracție (miopie, astigmatism)
- Glaucom/creșterea presiunii intraoculare
EDS miopatic (mEDS)
Mod de transmitere: autozomal recesiv sau autozomal dominant
Gena implicată: COL12A1
Pentru diagnosticul clinic al mEDS sunt necesare un criteriu major: hipotonie musculară congenitală care se îmbunătățește odată cu vârsta, plus un alt criteriu major și/sau trei criterii minore.
Criterii majore:
- Hipotonie musculară congenitală și/sau atrofie musculară, care se îmbunătățește odată cu vârsta
- Contracturi articulare proximale (genunchi, șold și cot)
- Hiperlaxitate a articulațiilor distale
Criterii minore:
- Piele moale, de consistență moale
- Cicatrizare atrofică
- Întârziere în dezvoltarea motorie
- Miopatie la biopsia musculară
EDS periodontal (pEDS)
Mod de transmitere: autozomal dominant
Genele implicate: C1R, C1S
Pentru diagnosticul clinic al pEDS sunt necesare criteriul major parodontită severă și refractară, cu debut timpuriu (în copilărie sau adolescență) sau criteriul major lipsa gingiei atașate plus cel puțin alte două criterii majore și un criteriu minor.
Criterii majore:
- Parodontită severă și refractară, cu debut timpuriu (în copilărie sau adolescență)
- Lipsa gingiei atașate
- Plăci pretibiale
- Istoric familial de rude de gradul întâi care îndeplinesc criteriile clinice
Criterii minore:
- Echimoze la traumatisme minime
- Hiperlaxitate articulară, în special la articulațiile distale
- Hiperextensibilitate și fragilitate a pielii, cicatrizare anormală (largă sau atrofică)
- Rată crescută a infecțiilor
- Hernii
- Trăsături faciale marfanoide
- Acrogerie
- Vasculatură proeminentă
Managementul și tratamentul sindromului Ehlers-Danlos
Strategii de gestionare a simptomelor
Abordări pentru gestionarea durerii, oboselii și problemelor de mobilitate
Gestionarea simptomelor în sindromul Ehlers-Danlos (EDS) necesită o abordare cuprinzătoare din cauza complexității afecțiunii. Durerea cronică este o problemă prevalentă, afectând până la 90% dintre pacienții cu EDS. Strategiile de gestionare a durerii includ terapia fizică, medicamente precum antiinflamatoarele nesteroidiene (AINS) și, în unele cazuri, opioide pentru dureri severe. Gestionarea oboselii implică adesea ajustări ale stilului de viață, inclusiv dozarea activităților și asigurarea unui repaus adecvat.
Recomandări pentru terapie fizică și terapie ocupațională
Terapia fizică este o piatră de temelie în gestionarea simptomelor EDS. Aceasta se concentrează pe întărirea mușchilor din jurul articulațiilor pentru a îmbunătăți stabilitatea, îmbunătățirea propriocepției pentru a preveni leziunile și gestionarea durerii prin exerciții și modalități personalizate. Terapia ocupațională poate fi, de asemenea, recomandată pentru a ajuta pacienții să adapteze activitățile zilnice și să îmbunătățească calitatea vieții. Tehnici precum terapia prin expunere la cald și rece, terapia manuală și exercițiile specifice de întindere sunt frecvent utilizate.
Intervenții medicale și chirurgicale
Când se ia în considerare intervenția chirurgicală pentru stabilizarea articulațiilor sau corectarea manifestărilor sau complicațiilor vasculare
Chirurgia este în general luată în considerare atunci când tratamentele conservatoare eșuează sau în cazuri de complicații severe, cum ar fi instabilitatea articulară sau urgențele vasculare. De exemplu, poate fi necesară intervenția chirurgicală pentru stabilizarea articulațiilor sau pentru a aborda problemele vasculare amenințătoare de viață în EDS vascular (vEDS).
Riscuri asociate intervențiilor chirurgicale la pacienții cu EDS
Pacienții cu EDS se confruntă cu riscuri crescute în timpul procedurilor chirurgicale din cauza fragilității tisulare. Aceste riscuri includ vindecarea lentă sau deficitară a plăgilor, sângerări excesive și complicații legate de anestezie. Prin urmare, intervențiile chirurgicale sunt abordate cu precauție, iar echipele chirurgicale specializate, familiarizate cu EDS, sunt adesea necesare.
Modificări ale stilului de viață și terapii de suport
Importanța schimbărilor de stil de viață și a strategiilor de gestionare zilnică
Modificările stilului de viață sunt cruciale pentru gestionarea EDS. Acestea includ ajustări ergonomice, încălțăminte de suport și alegeri dietetice care susțin sănătatea articulațiilor. Exercițiile fizice regulate, adecvate și tehnicile de gestionare a stresului, cum ar fi mindfulness și yoga, pot fi, de asemenea, benefice.
Rolul sprijinului pentru sănătatea mintală și consiliere
Sprijinul pentru sănătatea mintală este vital pentru pacienții cu EDS, care adesea se confruntă cu provocări psihologice din cauza durerii cronice și a restricțiilor de stil de viață. Consilierea și intervențiile psihologice, cum ar fi terapia cognitiv-comportamentală (CBT), pot ajuta pacienții să facă față poverii emoționale a bolii. Grupurile de sprijin, consilierea psihologică și consilierea genetică oferă, de asemenea, resurse valoroase pentru pacienți și familiile lor, ajutându-i să înțeleagă afecțiunea și să ia decizii informate despre îngrijirea lor.
În general, gestionarea EDS necesită o abordare multidisciplinară care să abordeze atât aspectele fizice, cât și cele psihologice ale sindromului, asigurând un plan de îngrijire cuprinzător adaptat nevoilor fiecărui pacient.
Viața cu sindromul Ehlers-Danlos – impactul asupra calității vieții
Sindromul Ehlers-Danlos (EDS) afectează semnificativ calitatea vieții, având un impact direct asupra funcționării zilnice, relațiilor și stării psihologice.
Durerea cronică, oboseala și limitările fizice sunt principalele simptome care influențează activitățile de zi cu zi și capacitatea de muncă. Mulți pacienți cu EDS au dificultăți în a-și menține un loc de muncă cu normă întreagă, din cauza cerințelor fizice și a imprevizibilității simptomelor. Durerile articulare și dislocările frecvente împiedică îndeplinirea sarcinilor obișnuite, ceea ce determină adaptări constante ale programului de muncă și reducerea activităților profesionale.
Relațiile interpersonale sunt, de asemenea, afectate, deoarece pacienții trebuie să se bazeze adesea pe sprijinul celor din jur și să își adapteze stilul de viață. Schimbările frecvente de planuri, necesitatea de a evita activități solicitante și dependența de ajutorul celor apropiați pot duce la tensiuni în relațiile sociale și familiale. În plus, retragerea din activitățile sociale din cauza durerii și a oboselii poate izola pacienții, accentuând impactul psihologic al afecțiunii.
Impactul emoțional este considerabil, mulți pacienți dezvoltând anxietate și depresie din cauza limitărilor impuse de boală. Natura cronică și imprevizibilă a EDS, alături de durerea constantă, contribuie la scăderea stimei de sine și la dificultăți în menținerea echilibrului emoțional. Sprijinul psihologic, inclusiv consilierea și terapia cognitiv-comportamentală, este esențial pentru a ajuta pacienții să gestioneze stresul și provocările emoționale asociate cu EDS.
Astfel, gestionarea EDS necesită nu doar intervenții medicale și terapeutice, ci și suport emoțional și adaptări constante ale stilului de viață pentru a îmbunătăți calitatea vieții pacienților.
Reproducerea persoanelor cu EDS
Persoanele cu EDS prezintă riscuri reproductive mai ridicate față de populația generală. Un aspect este riscul de a transmite afecțiunea copiilor, care poate fi de până la 50% pentru formele autozomal dominante.
Mai mult, a fost raportat faptul că femeile cu EDS suferă mai frecvent complicații legate de sarcină decât femeile neafectate, iar cele cu sindromul Ehlers-Danlos vascular pot suferi complicații amenințătoare de viață.
Complicații în timpul sarcinii
Femeile cu EDS, în special cele cu EDS hipermobil (hEDS) și vascular (vEDS) se confruntă cu riscuri mai mari pentru diverse complicații în timpul sarcinii, comparativ cu populația generală:
- Preeclampsie și eclampsie
- Ruptura prematură a membranelor
- Naștere prematură
- Hemoragie antepartum și postpartum
- Deces fetal intrauterin
- Hiperemezis gravidum
- Infecții ale plăgii cezariene
- Psihoză postpartum și tulburare de stres post-traumatic
Riscuri specifice EDS vascular
Pentru femeile cu EDS vascular (vEDS), sarcina și nașterea implică riscuri semnificative atât de morbiditate, cât și de mortalitate. Rata mortalității materne per sarcină pentru femeile cu vEDS este crescută semnificativ, fiind de aproximativ 5,7%, riscul de avort spontan este de aproximativ 13,8%, iar riscul de prematuritate este de aproximativ 8,8% [4].
Riscurile specifice includ:
- Ruptură uterină
- Evenimente vasculare: disecțiile vasculare sau rupturile de valve cardiace
- Evenimente digestive: perforarea organelor cavitare
Managementul sarcinii femeilor cu EDS
Femeile cu EDS necesită o monitorizare specializată multidisciplinară în timpul sarcinii asigurată de specialiști în obstetrică, medicină materno-fetală, cardiologie, ortopedie, genetică care să asigure managementul personalizat fiecărei paciente pentru evitarea riscurilor atât din timpul sarcinii cât și perinatale.
Prevenirea transmiterii afecțiunii către copii
Persoanele cu EDS prezintă un risc de a transmite afecțiunea copiilor. În cazul formelor cu transmitere autozomal dominantă, persoana cu EDS are o probabilitate de 50% de a transmite afecțiunea fiecăruia dintre copii. Iar în cazul persoanelor cu forme autozomal recesive, riscul de a avea un copil afectat este de 50% dacă și partenerul este purtător sănătos.
Consultația cu un medic specialist în genetică înainte de concepție va oferi informații despre riscurile reproductive, testele genetice potrivite pentru diagnosticul de certitudine al afecțiunii, testele potrivite pentru analiza partenerului reproductiv al persoanei afectate și testele genetice preconcepționale potrivite pentru prevenirea transmiterii afecțiunii copiilor sau pentru depistarea precoce.
Teste genetice prenatale pentru EDS
Testarea genetică a embrionilor în cadrul fertilizării in vitro
În cazul persoanelor cu risc crescut de a transmite EDS copiilor lor, pentru a împiedica trasmiterea afecțiunii se poate efectua fertilizare in vitro și testarea genetică a embrionilor înainte de implantare (PGT-M). Astfel, se poate asigura implantarea embrionilor neafectați și întrerupe transmiterea sindromului în descendență.
Testarea genetică a fătului în timpul sarcinii
În cazul în care sarcina este deja în curs, se poate efectua testarea genetică a fătului prin prelevarea invazivă a materialului genetic fetal prin biopsie de vilozități coriale sau amniocenteză.
Cercetare și direcții viitoare în sindromul Ehlers-Danlos
Cercetări actuale asupra sindromului Ehlers-Danlos
Rezumatul studiilor recente și concluziile acestora
Cercetările recente asupra sindromului Ehlers-Danlos (EDS) s-au concentrat pe aprofundarea cunoștințelor legate de etiologia și epidemiologia acestuia, precum și pe îmbunătățirea strategiilor de management clinic. Un accent deosebit a fost pus pe heterogenitatea EDS, în special pe tipul hipermobil (hEDS), pentru care încă nu s-a identificat o cauză moleculară clară. Studiile subliniază necesitatea unor cercetări extinse pentru a identifica biomarkeri genetici și moleculari, ceea ce ar permite un diagnostic mai precis și o înțelegere mai detaliată a patofiziologiei EDS. În plus, eforturile de cercetare continuă să dezvolte noi criterii de diagnostic, cu scopul de a reduce timpul până la diagnostic și de a elimina erorile frecvente care apar din cauza suprapunerii simptomelor cu alte afecțiuni ale țesutului conjunctiv.
Terapie emergentă și intervenții
O serie de abordări terapeutice emergente sunt în prezent studiate, aducând speranțe pentru îmbunătățirea calității vieții pacienților cu EDS. Printre acestea se numără gelul Excellagen, un tratament topic investigat pentru a accelera vindecarea rănilor, și diverse tehnici de medicină regenerativă, precum proloterapia, care promite îmbunătățiri în stabilizarea articulațiilor și reducerea durerii prin stimularea regenerării țesutului conjunctiv. În paralel, tratamentele inovative pentru gestionarea durerii, cum ar fi naltrexona în doze mici și modulatorii celulelor gliale, sunt supuse cercetărilor clinice pentru a evalua eficacitatea lor în gestionarea durerii cronice la pacienții cu EDS, în special în tipul hipermobil, unde durerea este adesea refractară la tratamentele convenționale.
Aceste cercetări reprezintă un pas important în găsirea unor soluții terapeutice mai eficiente, oferind speranțe pentru o gestionare mai bună a simptomelor și o reducere a impactului EDS asupra calității vieții.
Progrese în cercetarea genetică
Descoperiri noi în cercetarea genetică și implicațiile lor pentru EDS
Progresele recente în cercetarea genetică au dus la identificarea bazei moleculare pentru 12 din cele 13 tipuri recunoscute de sindrom Ehlers-Danlos (EDS), cu studii în desfășurare pentru a descoperi substratul genetic al EDS hipermobil (hEDS), care rămâne neelucidat. Utilizarea tehnologiilor de secvențiere de generație următoare (NGS) a extins semnificativ înțelegerea relațiilor genotip-fenotip în EDS, permițând astfel dezvoltarea unor criterii de diagnostic mai precise. Aceasta a deschis calea către strategii de management personalizate, bazate pe profilul genetic individual al pacienților, oferind oportunități pentru îmbunătățirea managementului simptomelor și reducerea complicațiilor.
Tratamente și terapii potențiale viitoare
Cercetările clinice actuale explorează noi direcții terapeutice pentru pacienții cu EDS. Un domeniu de interes este eficacitatea inhibitorilor factorului de creștere nervoasă, care pot oferi noi soluții pentru gestionarea durerii, în special în cazurile de durere cronică asociată cu hEDS. Aceste terapii emergente urmăresc să completeze strategiile tradiționale de gestionare a durerii, oferind pacienților opțiuni mai eficiente și mai puțin dependente de opioide.
În plus, Fundația de Cercetare pentru Sindromul Ehlers-Danlos susține activ cercetările axate pe intervenții de impact și terapii țintite, având ca obiectiv îmbunătățirea îngrijirii diagnostice și terapeutice. Proiectele susținute de fundație explorează potențialul unor terapii regeneratoare și modul în care acestea pot fi aplicate în stabilizarea țesuturilor conjunctive și în reducerea fragilității acestora. De asemenea, noile studii clinice și cercetările preclinice vizează o gamă largă de tratamente farmacologice care ar putea transforma gestionarea EDS, oferind pacienților speranța unor terapii mai eficiente în viitor.
În ansamblu, viitorul cercetării în EDS este promițător, cu studii în desfășurare care urmăresc să îmbunătățească atât înțelegerea mecanismelor moleculare ale bolii, cât și calitatea vieții pacienților prin soluții terapeutice mai bune și mai accesibile.
Concluzie
Sindromul Ehlers-Danlos (EDS) este o afecțiune genetică complexă, care afectează profund calitatea vieții pacienților prin manifestările sale variate și adesea severe. Cercetările recente oferă speranțe pentru îmbunătățirea diagnosticului și dezvoltarea de noi tratamente care să gestioneze mai eficient simptomele. Conștientizarea și înțelegerea acestei afecțiuni, atât de către profesioniștii din domeniul sănătății, cât și de către publicul larg, sunt esențiale pentru diagnosticarea precoce și pentru asigurarea unei îngrijiri adecvate și personalizate, care să reducă impactul bolii asupra vieții cotidiene.
Referințe:
- 1.Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B. 2017. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet Part C Semin Med Genet 175C: 8–26.
- 2.Parapia, L.A. and Jackson, C. (2008), Ehlers-Danlos syndrome – a historical review. British Journal of Haematology, 141: 32-35. https://doi.org/10.1111/j.1365-2141.2008.06994.x
- 3.https://www.ehlers-danlos.com
- 4.Haem T, Benson B, Dernoncourt A, Gondry J, Schmidt J, Foulon A. Vascular Ehlers-Danlos syndrome and pregnancy: A systematic review. BJOG. 2024 Nov;131(12):1620-1629. doi: 10.1111/1471-0528.17893. Epub 2024 Jun 26. PMID: 38926786.
Dr. Anca Neacșu, medic specialist în genetică medicală cu peste 10 ani de experiență, este expertă în consiliere genetică și interpretarea analizelor genetice avansate. Absolventă a Universității de Medicină și Farmacie “Carol Davila” din București și cu formări suplimentare la Harvard, Stanford și Asociația Americană de Nutriție, ea promovează o paradigmă medicală nouă, axată pe prevenție și tratamente personalizate o viață cât mai sănătoasă și prelungirea longevității.
Află mai multe despre Anca